关于mega4.0建系统进化树的若干问题
近日学习用mega4.0建系统进化树,大概会操作了,但仍然有很多疑问。坛子里关于mega的帖子看了遍,迷茫发问的朋友多,解答的大牛人也有,我的问题和很多求助网友相似,现系统提出,渴望知道的大侠尽早看到此贴,指点下迷津,不甚感激!
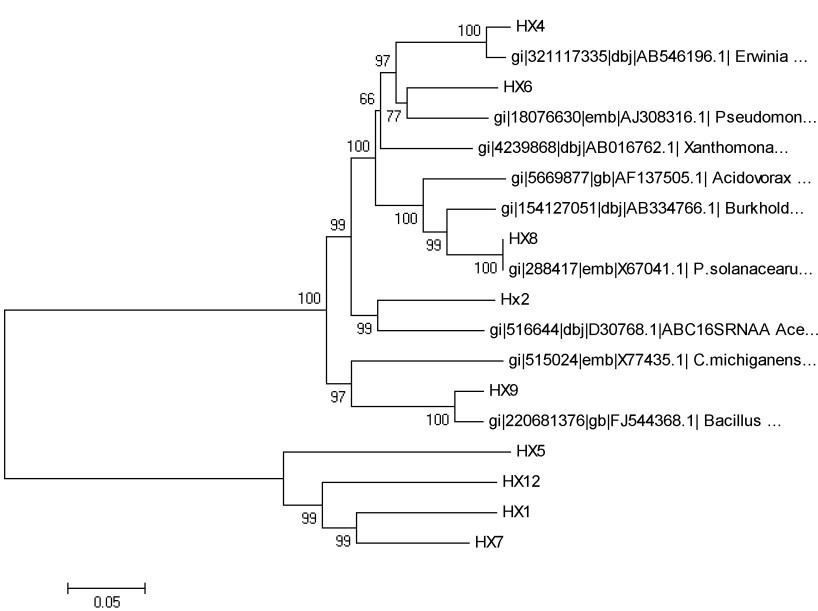
一,附件中的小树是用mega4.0,Bootsrap Value设置1000次建成。具体步骤如下:
a、打开mega软件—Alignment 下拉—Alignment exposure/clustal—create a new alignment --然后根据实际情况(DNA or 蛋白序列)选择Yes
b、Edit—Insert sequence from file(.fastal的格式);
c、Algin by clustal W---Data—Export Alignment—Mega fomat;
d、打开上一步保存的文件—Distance—compute pairwise—compute;
e、Phylogeny—Bootstrap Test of phylogeny—Neighbor-Joining—将Boostrap(500 replicates;seed=64238修改为1000 replicates)--compute---获得进化树—设好格式---image –save as TIF file
问题来了,在坛子里看见各位大侠给出的步骤,要用Clustal X软件进行序列比对之后才能建树,请问,mega4.0中是不是嵌合了clustal么,为什么有大侠说还要下载一个clustal软件呢?上面的建树步骤C是不是算经过了clustal呢?
二,附件中小树节点处数值的含义,请各位解释下为什么HX4和AB546196.1(bootstrap值100)与HX6和AJ308316.1(bootstrap值77)两类群聚一起后又97呢?是否意味着HX4和AB546196.1亲缘关系很近,甚至是同一个东西,HX6和AJ308316.1关系较近,这两类群又聚一起,而且bootstrap值97,大于77,很费解呀很费解。
三,树下的标尺有什么用?去算枝长吗?枝长又代表什么,进化距离吗?如果枝长是进化距离,那bootstrap值又代表什么?
四,在坛子里逛的时候,看到有大侠说“遗传标度法和步长值法是用来表示两种不同的结果时用的 步长值常用在鉴定菌种的进化树中 而遗传距离法常用在表示不同菌株间进化关系的进化树中 ”。什么是遗传标度法,什么是步长值法?本人建树目的是想得到菌株间的进化关系
五,看文献过程中,发现有部分文献的进化树要么只有标尺,要么只有bootstrap值,而且也没有对数值,标尺的含义做出解释,究竟什么样的树才算规范?
昨晚码了一个求助贴,满怀希望发的时候,系统维护,伤啊伤!今天抖擞一下,再次情绪饱满的发一个,敬候各位的佳音哈!
1、clustalx 也是为了alignment,如果在MEGA里面alignment了,就不用clustalx了;
2、bootstrap值一般是将你的序列保留一部分,把剩下部分随机打乱,拼成不同的序列,组成1000个你的alignment文件,做树,显示的77表明,1000次做树的过程有有77%次,也就是770次得到HX6和AJ308316.1聚在一起这个结果;
3、下面的标尺是枝长,也是进化距离,bootstrap值是可信度;
4、这个我水平有限,也比较迷惑,个人觉得你现在做成的树适合的进化研究;
5、一般是要有bootstrap值的,这个表示可信度,要是值低于50,一般别人是不认同的,有的文章没有标尺,可能是只想得到树的分枝情况,没有想要计算各个物种的距离,只要得到的一个树的拓扑结果吧,但是bootstrap值一般是要的。
对了,你的alignment,排列好了之后,一般要将首尾序列截成一样齐……
只是个人意见,可能有偏差,有错误欢迎指出。
1、为什么标尺有时候是1,2,有时候是0.05,0.02?如何设置标尺大小?
2、标尺用来算遗传距离,具体怎么算呢(比如,在附件的图中HX4与AB546196这一群和HX6与AJ308316这一群的距离看哪根树枝呢?或者不相邻的,再远一点儿的类群要看它们之间的进化距离怎么看呢?)
或者phylip软件里面的dnadist,我觉得如果不是做进化之类的分析的话,一个diversity distance 就可以吧,mage里面好像就有
不过个人感觉有时候截不截影响不是太大
刚试了个一百多个数据的比对 有不齐的 和剪切后的
两种的树做出来一样
不过我是做的蛋白树 不知道金银的会怎样=。=
我觉得貌似BOOTSTRAP值这个可信度是不是有待商榷
我做的时候跟老板商量的据说是75以下的都容易变
这个变是只插入或删除别的序列后容易摆动
所以具体这个数值是多少我也没查文献
看很多人说50 我觉得对此还是根据自己不同情况分析以下比较好
bootstrap值低于50确定是不可信的,但高于50只能说针对当前数据,在bootstrap方法检验下,是高于一半的支持率的,这个值当然越高越好,你选择不同数据,不同模型,bootstrap值一般都会有变化的,bootstrap值为100%也只能说是两个序列聚在一起的概率非常大,是个推测值,也不能绝对说他们就是一类的,我个人觉得就是一个统计分析结果,低于50%一般是不可信,高于50%可信度就高一点而已……
我还做了一个别的实验
发现有些东西剪切掉了对树是有非常大的影响的 比如说一些LINKER
关于BOOSTRAP值 我明白你意思 我的意思不是问这个变化怎样 我的意思是这个值是不是应该调到75=。=b
那不同的物种序列长度肯定是有差异的做作树时,软件会自己处理这些长度差异的问题吧!
今年刚接触分子系统学,当时老师上课时说序列不用截齐!